
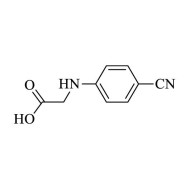
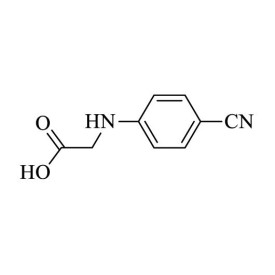
Extensive quantum chemical calculation have been carried out to investigate the Fourier Transform Infrared(FT-IR), Fourier Transform Raman(FT-RAMAN) and Nuclear magnetic resonance(NMR), and Ultra Violet-Visible(UV-vis) spectra of 2-(4-Cyanophenylamino) acetic acid. The molecular structure, fundamental vibrational frequencies and intensities of the vibrational bands were interpreted with the aid of optimizations and normal coordinate force field calculations based on density functional theory (DFT) and ab initio HF methods with 6–311++G(d,p) basis set. The theoretical vibrational wavenumbers are compared with the experimental values. The calculated HOMO-LUMO energies were found to be-6.2056 eV and -1.2901 eV which indicates the charge transfer within the molecule. Natural bond orbital analysis has been carried out to explain the charge transfer (or) delocalization of charge due to the intra molecular interactions. Molecular Electrostatic Potential (MEP), First order hyperpolarizability, and Fukui functions calculation were also performed. The thermodynamic properties of the title compound were studied for different temperatures. Molecular docking studies were made on the title compound to study the hydrogen bond interactions and the minimum binding energy was calculated.